
EURL
European Union Reference Laboratory for
Brucellosis

The present document describes a standard technique aiming at controlling the fulfilment of OIE and EU requirements regarding the standardisation of diagnostic antigens for the detection of antibodies specific of smooth Brucella species (especially B. abortus, B. melitensis and B. suis):
- in animal individual sera by the Rose Bengal Test (RBT) and the Complement Fixation Test (CFT);
- in bovine pooled milk samples by the Milk Ring-Test (MRT).
The present document describes a standard technique aiming at detecting antibodies specific of Brucella ovis by the complement fixation test in ovine sera.
The present document describes a standard technique that allows establishing species and biovars of an identified Brucella culture. Species identification is based on two main sets of properties: lysis by phages and oxidative metabolic profiles on selected amino acid and carbohydrate substrates. The described identification techniques enable to establish species and biovar of specific Brucella culture.
The present document describes a standard technique that allows research and bacteriological identification of the Brucella genus, in animal samples (ruminants, equidae, suidae, camelidae and carnivores, marine mammals, both wild and domestic). This is an isolation technique on solid culture agar medium. The described identification techniques enable a presumptive identification of bacteria of the Brucella genus.
The Bruce Ladder (BL) and the Bruce Ladder Suis (BLS) are two molecular typing tools designed to identify Brucella species for BL (Garci-Yoldi et al., 2006 ; Lopez-Goñi et al., 2008 ; Lopez-Goñi et al., 2011) and to differentiate Brucella suis biovars, Brucella canis and Brucella microti for the BLS (Lopez-Goňi et al. 2011 ; OIE Terrestrial Manual 2018, Chapter 3.1.4). These methods are applied on DNA extracted from calibrated suspensions of isolated strains.
Principle
Both methods consist in the separation of different DNA fragment by electrophoresis previously amplified by a multiplex Polymerase Chain Reaction.
A PCR multiplex is a method allowing the amplification of several distinct DNA fragments in one reaction, with all primers related to the amplification of different loci introduced into the same reaction tube.
Primers used for the PCRs are:

Once amplified, the DNA fragments are deposited on an agarose gel and are separated according to their size by electrophoretic migration
Interpretation
After migration, a picture is taken and the DNA fragments of samples tested are compared with the fragments of reference strains (used as controls).

The Real Time Polymerase Chain Reaction (PCR) or Quantitative PCR is a molecular diagnostic tool used to detect nucleic acids, and in this case Brucella DNA (Bounaadja et al., 2009).
Principle
The present document describes a standard technique aiming at real-time Polymerase Chain Reaction (PCR) to detect the presence / absence of Brucella DNA in animal / human samples. Prior to PCR, the DNA extraction protocols have to be validated for each type of sample handled in the laboratory in order to assure standardized performances. The ID Gene® Brucella spp Triplex (IDBRU) kit is a Reel-time Polymerase Chain Reaction (Rt-PCR) kit for specific Brucella spp. sequence amplification based on TaqMan® technology. This is a qualitative Triplex system that enables simultaneous amplification of the target DNA and internal endogenous and exogenous controls.
Scientific references
HOLZAPFEL M., 2018. De l’épidémiologie moléculaire aux analyses fonctionnelles de Brucella chez les ruminants, une approche intégrée pour l’identification et l’étude de la diversité phénotypique d’un genre génétiquement homogène. Thèse de doctorat en Microbiologie, soutenue le 26-11-2018 à Paris Est, dans le cadre de l’École doctorale ABIES, 200 pages. Online document: http://www.theses.fr/2018PESC1141/document
An ELISA is a plate-based laboratory technique making use of the binding between an antigen and its homologous antibody in order to identify and quantify the specific antigen or antibody in a sample. ELISAs are typically performed in 96-well (or 384-well) polystyrene plates, which will passively bind antibodies and proteins.
c-ELISA kits have to be previously validated according to EURL specifications. c-ELISA tests have to be performed according to manufacturer instructions.
INTRODUCTION
Antibodies also known as immunoglobulins (Ig) are gamma globulin proteins that are found in blood. These specialized immune proteins are produced following the introduction of an antigen into the body and possess the remarquable ability to combine with the antigen that triggered its production (specific affinity). The antibody recognises and bind to the antigenic determinant region of the antigen.
Enzyme-linked immunosorbent assays rely on specific antibodies to bind the target antigen, and a detection system to indicate the presence and quantity of antigen binding.
Brucella antigens (for review, Ducrotoy et al., 2016, DOI: 10.1016/j.vetimm.2016.02.002).
Like other gram negative bacteria, Brucella consist of cytoplasm surrounded by a cell envelope made of an inner membrane, periplasm and an outer membraine. The outer membrane contains two main classes of antigens: lipopolysaccharide (LPS) and detergent-soluble proteins (referred to as outer membrane proteins [Omp]). Structurally, the LPS can be either smooth (S) or rough (R) depending on the Brucella species. These two LPS differ in the presence in the former and absence in the latter of a long polysaccharide section, the O-polysaccharide
The antigens relevant to ruminant brucellosis can be classified into 2 categories: S-LPS and the related haptenic polysaccharides (O-polysaccharide linked with most core oligosaccharide sugars or core-O-polysaccharides ; the native haptens (NH) and the polysaccharide B) ; and protein antigens. The O-polysaccharide carries three basic antibody reactivities: A, M and C (Douglas and Palmer, 1988). In diagnosis, the criticial concept is the existence in the serum of an infected animal of a range of antibody reactivities (A>M, A= M and A<M) that in practice correspond to overlapping epitopes highly repeated in the O-polysaccharide (hence their colloective name as Brucella C epitope ; Weynants et al., 1997). Brucella O-polysaccharides cross-react with gram-negative bacteria that contain variable proportions of N-substituted perosamine. Among these bacteria, Yersinia enterocolitica serotype O:9 generates the strongest cross-reactivity.
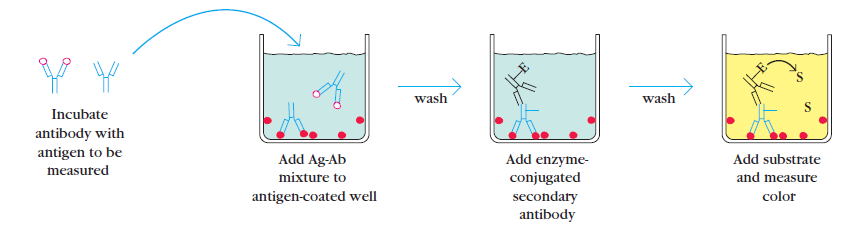
Principle of C-ELISA (source: Laboratoryinfo.com)
This type of ELISA depends on the competitive reaction between the sample antigen and antigen bound to the wells of microtiter plate with the primary antibody.
1-First, the primary antibody is incubated with the sample. This results in the formation of Ag-Ab complex which are then added to the wells that have been coated with the same antigens.
2-After an incubation, unbound antibodies are washed off. The more antigen in the sample, more primary antibody will bind to the sample antigen. Therefore there will be smaller amount of primary antibody available to bind to the antigen coated on well.
3-Secondary antibody conjugated to an enzyme is added, followed by a substrate to elicit a chromogenic signal.
Concentration of color is inversely proportional to the amount of antigen present in the sample.
Before starting the work, read kit instruction carefully!
Results
The ELISA assay yields three different types of data output:
Serological tests for Brucellosis [relayed from OIE Terrestrial Manual]:
The buffered Brucella antigen tests (rose bengal test and buffered plate agglutination test), the complement fixation test, the enzyme-linked immunosorbent assays (ELISA) or the fluorescence polarisation assay, are suitable tests for screening of herds/flocks and individual small ruminants, camelids and bovines (cattle and buffaloes). However, no single serological test is appropriate in each animal species and all epidemiological situations, and some of these tests are not adequate for diagnosing brucellosis in pigs. Therefore, the reactivity of samples that are positive in screening tests should be assessed using an established confirmatory or complementary strategy. The indirect ELISA or milk ring test performed on bulk milk samples is effective for screening and monitoring dairy cattle.
Several variations of the C-ELISA, using S-LPS or O-PS as antigens, have been described for cattle, small ruminants and pigs employing different antiglobulin-enzume conjugates, substrate or chromogens and antigens prepared from different smooth Brucella strains. Nevertheless, the technique used and the interpretation of results must have been validated in accordance with OIE requirements. It has been shown that the C-ELISA eliminates some but not all False Positive Serological Reactions (FPSR) caused by cross-reacting bacteria in cattle (Muñoz et al., 2005) and swine (Praud et al., 2012). In some cases, in ruminants or pigs, FSPR may be observed in C-ELISA while not in other S-LPS-based tests, including I-ELISA. Moreover, the C-ELISA reduces but not fully eliminates the reactions caused by antibodies produces in response to vaccination.
Some protocols are less sensitive or less specific than others; therefore results obtained from different assays are not always comparable. C-ELISA for diagnosing anti-Brucella antibodies in small ruminants and swine is essentially te same as that described for cattle, but the cut-off should have been properly establhised for these species using the appropriate validation techniqes.
Whatever the C-ELISA format used:
i) a positive and a negative control are included in each plate. Optical Density (OD) ranges to be obtained with these two controls must be established to define the criteria for validating each plate results. The OD of the positive control is the one with which the OD of each test serum is compared to establish the final result -negative of positive).
ii) an additional positive serum (internal control) must be included in each plate to validate the repeatability of the test from plate to plate and from day to day (control chart).
The complement fixation test (CFT) is used to detect the presence of specific antibodies in the patient’s serum. This test is based on the use of complement, a Biologically labile serum factor that causes the immune cytolysis i.e. lysis of antibody coated cells.
Principle (source Laboratoryinfo.com)
It is the nature of the complement to be activated when there is formation of antigen-antibody complex.
The first step is to heat the serum at 56°C to destroy patient’s complement.
A measured amount of complement and antigen are then added to the serum.
If there is presence of antibody in the serum, the complement is fixed due to the formation of Ag-Ab complex. If no antibody is present then the complement remains free.
To determine whether the complement has been fixed, sheep RBCs and antibodies against sheep RBCs are added.
Interpretation
Positive test : The available complement is fixed by Ag-Ab complex and no hemolysis of sheep RBCs occurs. So the test is positive for presence of antibodies.
Negative test : No Ag-Ab reaction occurs and the complement is free. This free complement binds to the complex of sheep RBC and it’s antibody to cause hemolysis, causing the development of pink color.
An ELISA is a plate-based laboratory technique making use of the binding between an antigen and its homologous antibody in order to identify and quantify the specific antigen or antibody in a sample. ELISAs are typically performed in 96-well (or 384-well) polystyrene plates, which will passively bind antibodies and proteins.
i-ELISA kits have to be validated according to EURL spectifications. i-ELISA tests have to be performed according to manufacturer instructions.
INTRODUCTION
Antibodies also known as immunoglobulins (Ig) are gamma globulin proteins that are found in blood. These specialized immune proteins are produced following the introduction of an antigen into the body and possess the remarquable ability to combine with the antigen that triggered its production (specific affinity). The antibody recognises and bind to the antigenic determinant region of the antigen.
Enzyme-linked immunosorbent assays rely on specific antibodies to bind the target antigen, and a detection system to indicate the presence and quantity of antigen binding.
Brucella antigens (for review, Ducrotoy et al., 2016, DOI: 10.1016/j.vetimm.2016.02.002).
Like other gram negative bacteria, Brucella consist of cytoplasm surrounded by a cell envelope made of an inner membrane, periplasm and an outer membraine. The outer membrane contains two main classes of antigens: lipopolysaccharide (LPS) and detergent-soluble proteins (referred to as outer membrane proteins [Omp]). Structurally, the LPS can be either smooth (S) or rough (R) depending on the Brucella species. These two LPS differ in the presence in the former and absence in the latter of a long polysaccharide section, the O-polysaccharide.
The antigens relevant to ruminant brucellosis can be classified into 2 categories: S-LPS and the related haptenic polysaccharides (O-polysaccharide linked with most core oligosaccharide sugars or core-O-polysaccharides ; the native haptens (NH) and the polysaccharide B) ; and protein antigens. The O-polysaccharide (O-PS) carries three basic antibody reactivities: A, M and C (Douglas and Palmer, 1988). In diagnosis, the criticial concept is the existence in the serum of an infected animal of a range of antibody reactivities (A>M, A= M and A<M) that in practice correspond to overlapping epitopes highly repeated in the O-polysaccharide (hence their colloective name as Brucella C epitope ; Weynants et al., 1997). Brucella O-polysaccharides cross-react with gram-negative bacteria that contain variable proportions of N-substituted perosamine. Among these bacteria, Yersinia enterocolitica serotype O:9 generates the strongest cross-reactivity.
Principle of I-ELISA (source: Laboratoryinfo.com)
1-Coating: the protein antigen to be tested for is added to each well of ELISA plate, where it is given time to adhere to the plastic through charge interactions.
2-Blocking: a solution of non-reacting protein is added to block any plastic surface in the well that remains uncoated by the protein antigen.
3-Then the serum is added, which contains a mixture of the serum antibodies, of unknown concentration, some of which may bind specifically to the test antigen that is coating the well.
4-Detection: afterwards, a secondary enzyme-conjugated antibody is added, which will bind to the antibody bound to the test antigen in the well. The secondary antibody is usually an anti-species antibody and is often polyclonal. A number of enzymes have been employed for ELISA, including alkaline phosphatase, horseradish peroxidase, and B-galactosidase.
5-Reading results: a substrate for this enzyme is then added. Often, this substrate changes colour upon reaction with the enzyme. The colour change shows that secondary antibody has bound to primary antibody, which strongly implies that the donor has had an immune reaction to the test antigen.
The higher the concentration of the primary antibody that was present in the serum, the stronger the colour change. Often a spectrometer is used to give quantitative values for colour strength.
Before starting the work, read kit instruction carefully!

Results
The ELISA assay yields three different types of data output:
Serological tests for Brucellosis [relayed from OIE Terrestrial Manuel]:
The buffered Brucella antigen tests (BBATs; rose bengal test and buffered plate agglutination test), the complement fixation test, the enzyme-linked immunosorbent assays (ELISA) or the fluorescence polarisation assay, are suitable tests for screening of herds/flocks and individual small ruminants, camelids and bovines (cattle and buffaloes). However, no single serological test is appropriate in each animal species and all epidemiological situations, and some of these tests are not adequate for diagnosing brucellosis in pigs. Therefore, the reactivity of samples that are positive in screening tests should be assessed using an established confirmatory or complementary strategy. The indirect ELISA or milk ring test performed on bulk milk samples is effective for screening and monitoring dairy cattle.
The I-ELISAs that use S-LPS or O-PS as antigens are highly sensitive for the detection of anti-Brucella antibodies in cattle, small ruminants and pigs, but are not capable of fully resolving the problem of differentiating between antibodies resulting from B. abortus S19 and B. melitensis Rev.1 vaccination. The B. abortus RB51 may also interfere in S-LPS-based I-ELISAs. Using I-ELISA standardised against the OIE standard sera, the diagnostic sensitivity should be equal or greater than that of the BBATs or the CFT (complement fixation test) in the testing of infected cattle, small ruminants and pigs. However, the specificity should be lower (Praud et al., 2012). The problem of False Positive Serological Reactions (FPSR) may be reduced but not fully resolved, in pigs in particular, by performing I-ELISAs using extracts from rough strains of Brucella. Most FPSR are a result of cross reaction with the O-PS portion of the S-LPS molecule; cross-reaction amoung core epitopes is less frequent but does exist.
Several commercial I-ELISAs are available. Some protocols are less sensitive or less specific than others; therefore results obtained from different assays are not always comparable. I-ELISA for diagnosing anti-Brucella antibodies in small ruminants and pigs is essentially the same as that described for cattle, but the cut-off should have been properly established for these species using the appropriate validation techniques, and moreover, I-ELISA for sheep and goats should be standardised against the ISaBmS (McGiven et al., 2011).
Whatever the I-ELISA format used:
i) a positive and a negative control are included in each plate. Optical Density (OD) ranges to be obtained with these two controls must be established to define the criteria for validating each plate results. The OD of the positive control is the one with which the OD of each test serum is compared to establish the final result -negative of positive).
ii) an additional positive serum (internal control) must be included in each plate to validate the repeatability of the test from plate to plate and from day to day (control chart).
Multiple Locus Variable number of tandem repeats Analysis (MLVA) is an assay based on length variation in tandem repeats of genomes. These markers (microsatellites, repeat units up to 8 bp, and minisatellites) can easily be tested for polymorphism and MLVA is one of the most widely used strategy for molecular typing of pathogenic bacteria.
The first MLVA Brucella assay was named "HOOF-prints" (Bricker et al., 2003). Eight highly variable loci present in most Brucella species were selected. These markers offered a very high discriminatory power (useful for outbreak investigations) but cannot provide a species assignment, especially due to a high level of homoplasy.
Other schemes have then been challenged and developed. The most widely used is the commonly named MLVA-16 (Le Flèche et al., 2006). This assay is divided into 2 different panels, one with a low discriminatory power (quick assignment to a species) and another one with a high discrimatory power (outbreak investigations).
Specific schemes related to species are regularly proposed, for example one scheme dedicated to B. suis biovar 2 named MLVA-11suis2 (Munoz et al., 2019).
MLVA for Brucella is a highly powerful tool for a rapid molecular identification. Several studies are performed on a limited number of strains, but one of the main point of this method is the comparison of the results between laboratories: a public database exist and a quick comparison of the results is easy, as results are a simple numeric code. More larger studies are allowed and confirm the accuracy of MLVA-16 for Brucella typing (Vergnaud et al., 2018). Nevertheless, even MLVA is efficient for strains clustering, phylogeny can not be precisely defined, especially due to rapidly evolving markers that can suffer from hompolasy.